Highlighted Publications
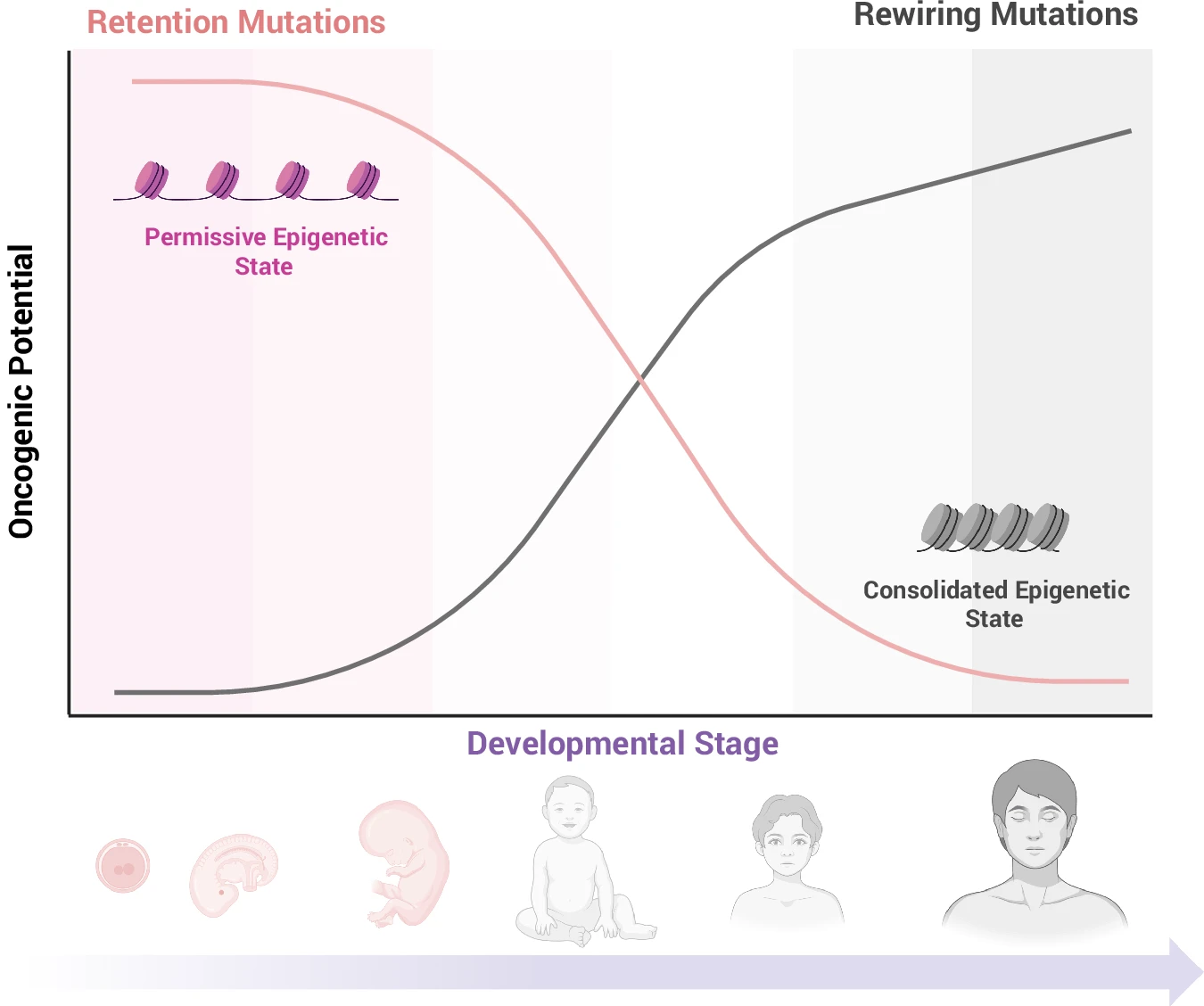
Developmental timing distinguishes pediatric and adult cancers through retention and rewiring mechanisms.
Nature Communications, Jul 03, 2026
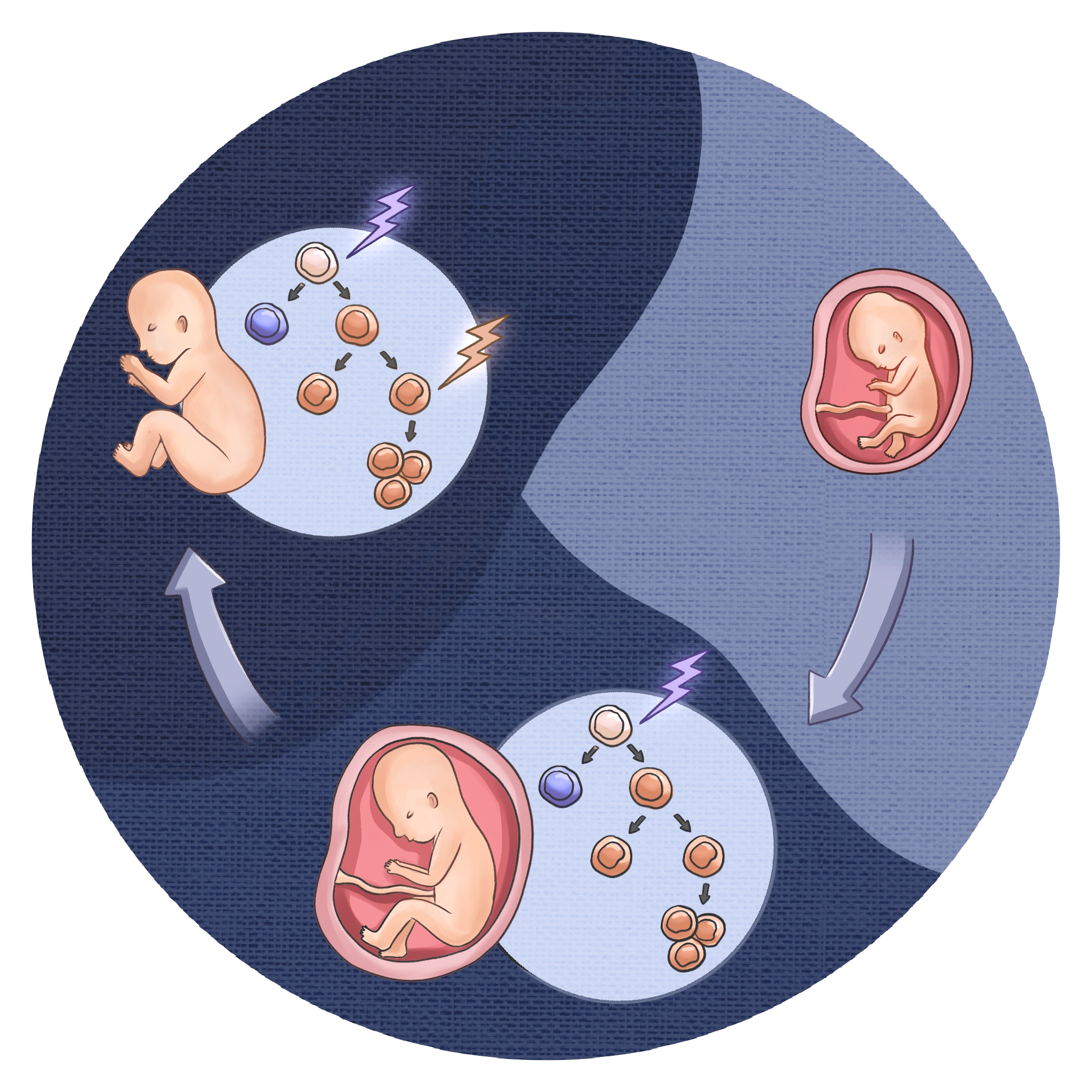
In this Perspective, we propose a unifying model in which developmental timing determines how the same oncogenic mutation shapes pediatric versus adult cancers. We distinguish early-life “retention” lesions, which stabilize already-active embryo-fetal transcriptional and epigenetic programs with few additional hits, from later-life “rewiring” lesions, which reactivate silenced developmental states through stepwise transcriptional and chromatin remodeling. Drawing on neuroblastoma, AML, and B-ALL, we argue that the epigenetic state of the cell of origin, not chronological age alone, dictates lineage trajectory, clinical behavior, and therapy response, motivating the integration of developmental timing into cancer risk stratification and treatment.
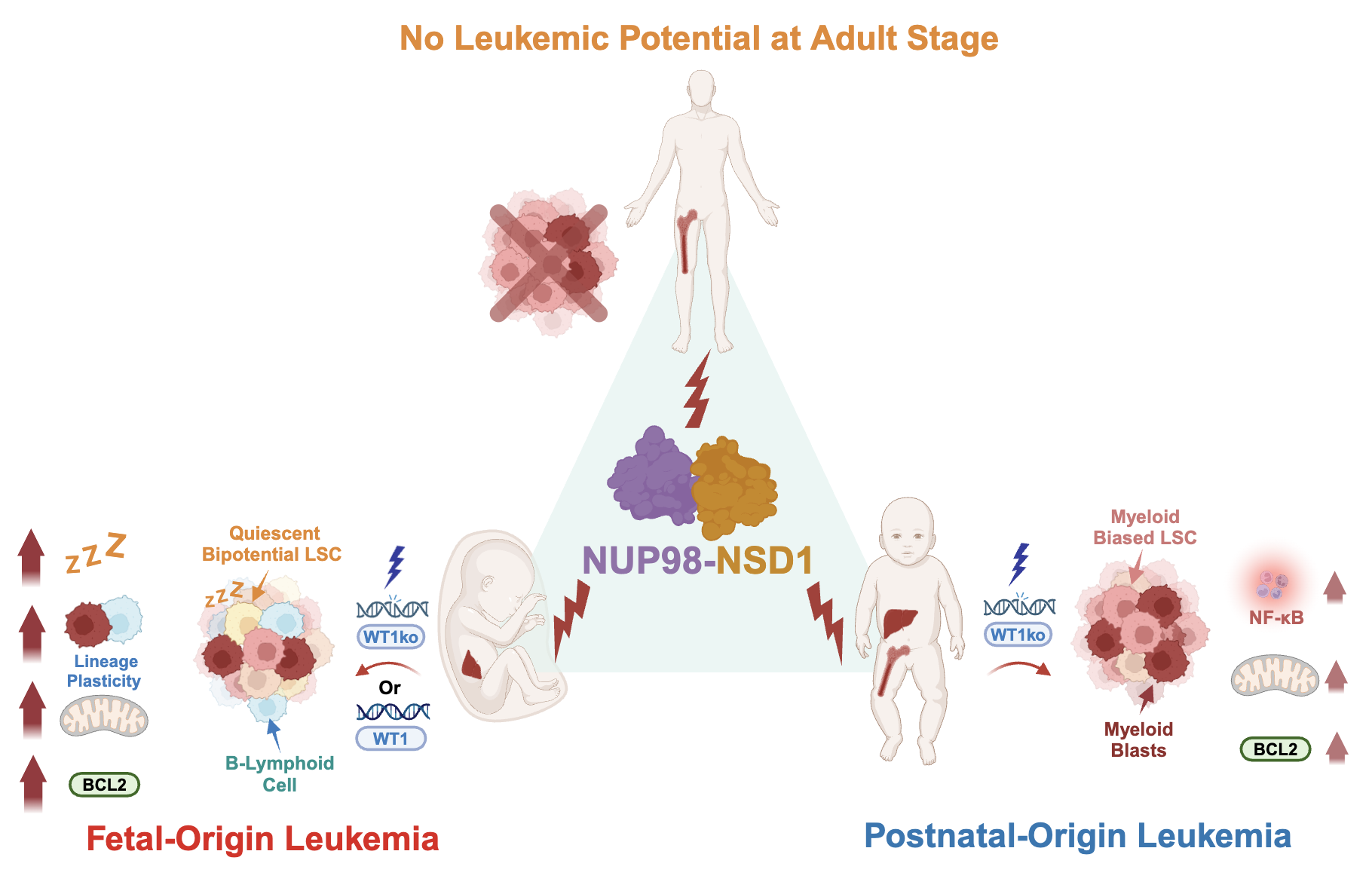
Ontogeny Dictates Oncogenic Potential, Lineage Hierarchy, and Therapy Response in Pediatric Leukemia
Cancer Discovery, Mar 02, 2026
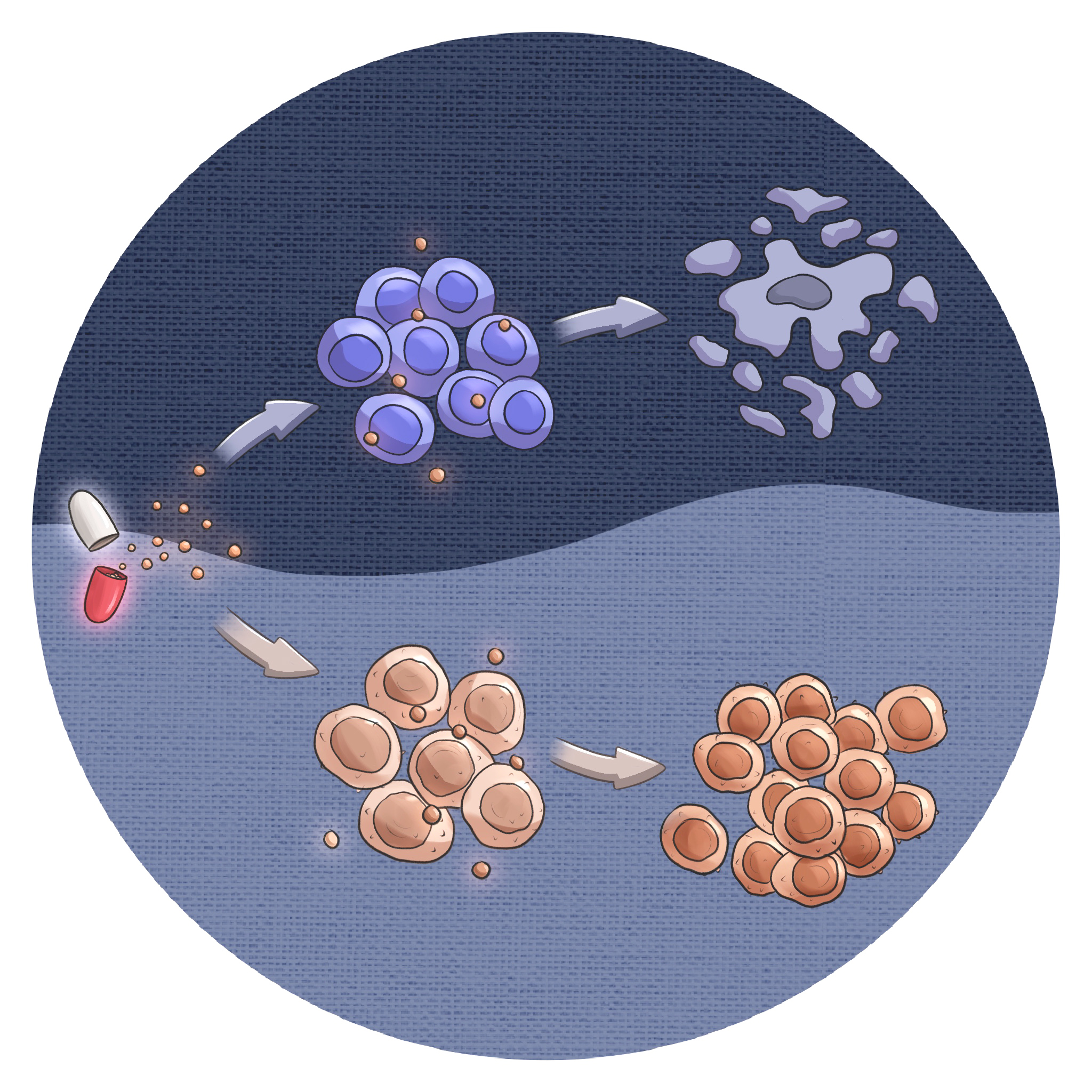
In this recently published study, we investigated the role of the NUP98-NSD1 fusion oncogene in pediatric acute myeloid leukemia (AML), demonstrating that its expression in human hematopoietic stem and progenitor cells leads to leukemic transformation. We further elucidated that the oncogenic potential of NUP98-NSD1 is influenced by the ontogeny of the cells, highlighting the importance of developmental context in leukemia pathogenesis.
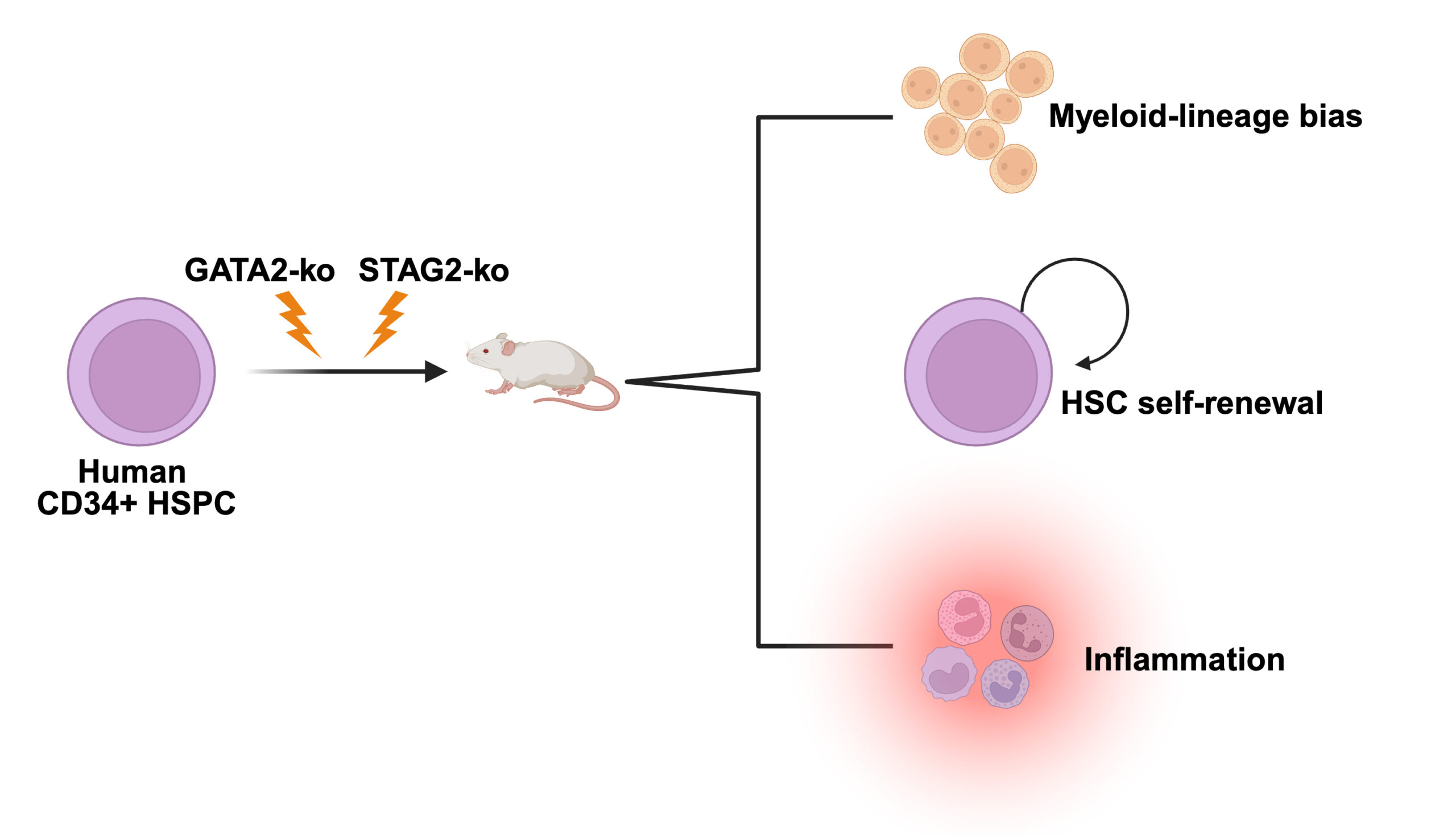
A CRISPR-Based Humanized Model Reveals Cooperative Role of STAG2 Loss in Familial GATA2-Deficient MDS Progression
bioRxiv [Preprint], Feb 02, 2026
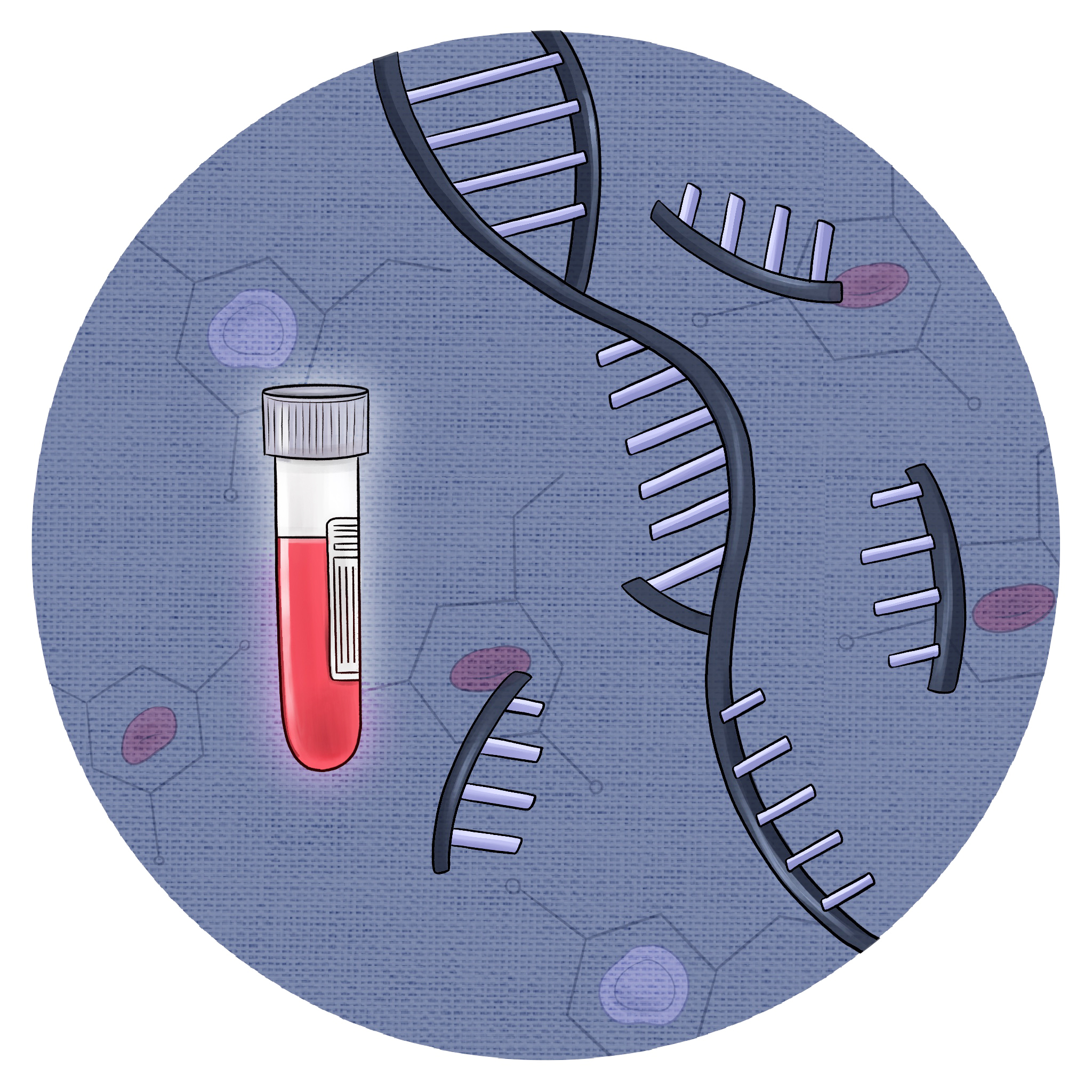
In this preprint, we developed a CRISPR/Cas9-engineered humanized model of familial GATA2-deficient myelodysplastic syndrome (MDS) to define how STAG2 loss cooperates in disease progression. Genome editing of primary human hematopoietic stem and progenitor cells followed by xenotransplantation demonstrates that STAG2 loss enhances stem cell persistence and myeloid skewing in the context of GATA2 deficiency. Single-cell analyses identify transcriptional programs linked to stemness and inflammatory signaling, providing mechanistic insight into high-risk GATA2-mutant MDS evolution.